
This is the first of a new series of posts where we highlight recent developments in laminin research.
In the first few months of 2018 there have been a little flurry of papers showing new or improved potential for therapies for inherited diseases where the mutation affects laminin function. So I thought I would group them together and do a little recap. Each of these studies have come at the problem of how to fix the defective laminins in different ways; full protein therapy, gene therapy and chimeric proteins. They are all impressive in their own right, building on a wealth of previous work and each could ultimately make a big difference to patient quality of life.
Laminin diseases?
Very quick intro (scroll down if you know about laminins already!). Laminins are key constituents of structures called basement membranes that are the foundations upon which sheets of cells sit. Basement membranes provide critical mechanical support to tissues as well as serving a plethora of other functions such as controlling the passage of molecules across the tissue (you can read our introduction to basement membranes here or about laminins here).
There are 12 different laminin genes – 5 alpha, 4 beta and 3 gamma – with each laminin being a trimeric protein consisting of one alpha, one beta and one gamma chain (eg LM521 consists of laminin α5, β2 and ϒ1). Different tissues have their own specific requirements for how much of each laminin they produce/need for the different activities that their tissue supports. Inherited mutations in specific laminin genes are therefore associated with different diseases depending on where that laminin is found and how crucial it is to that tissue’s function.
The diseases described here are a form of muscular dystrophy called merosin deficient congenital muscular atrophy, which occurs due to mutations in the LAMA2 gene, and a kidney/ocular disease called Pierson syndrome, which occurs due to mutations in LAMB2. Importantly, some of the approaches being developed might also work for some of the other laminin diseases.
Full Protein Therapy
Laminin-521 Protein Therapy for Glomerular Basement Membrane and Podocyte Abnormalities in a Model of Pierson Syndrome. by Lin et al. (paywall) in the Journal of American Society of Nephrology

What’s the approach? The authors injected fully assembled LM521 (~800kDa) intravenously into Pierson syndrome (lamb2-deficient) mice. The injected protein lined the glomerular basement membrane in the appropriate orientation and the mice got better; they showed reduced expression of proteins associated with injury and delayed the onset of proteinuria. However, it did not prevent nephrotic syndrome.
What’s next? This paper is really encouraging in that they added a huge protein to the bloodstream of the animals and were able to repair a specific kidney function. Although this LM521 treatment wouldn’t remove all the aspects of the disease, it does address the biggest contributor to poor quality of life for the patients so it looks promising.
The glomerular basement membrane is a pretty complex structure so that this works at all is somewhat surprising! Having said that, the idea of a large matrix protein being exogenously added has been shown to work for Collagen VII in dystrophic epidermolysis in 2004 intradermally (paper) andin 2013 for the blood stream (paper) .
Hurdles to overcome to push this forward would include challenges associated with production scale, purity and stability of the protein therapy, and the small number of patients with the disease means that big Pharma might not be interested. There is also a question about whether the exogenously added protein would be recognised as foreign and therefore raise an immune response. However, i.v. LM521 might help not only Pierson syndrome patients but also other kidney/glomerulus disorders where BM dysfunction occurs or for other kidney filtration disorders. One of the other good things is that the laminin proteins are quite long-lived with slow turn over rates so a protein therapy may not need to be injected very frequently to have substantial effects.
Gene Therapy
Laminin α1 reduces muscular dystrophy in dy2J mice by Gawlick et al published in Matrix Biology
What’s the approach? In these studies (this is a follow-up from some of the same group’s previous work), the authors have tried to rescue/prevent a disease state from occurring, not by adding back the missing protein but rather by inducing increased expression of a different laminin subunit in order to compensate for the one that is missing. Previously this group had shown that this works well where the mutation means that none of the laminin alpha2 protein is made, and they induce expression of laminin alpha1 on this background. In this most recent paper, they have used mice models where the mutation instead means that the mice still make some laminin alpha2 protein but the protein produced lacks its N terminus. This leads to a milder form of the disease that presents later in life.
Despite the disease being milder, the prediction was that their therapy might not work. Because laminins are obligate heterotrimers, the excess laminin alpha1 in this model presumably competes for the same beta and gamma chains as the mutant laminin alpha2 protein. In this case you might predict to end up with a mix of mutant and non-mutant laminins in the basement membrane. Importantly, the part of the protein that is missing in the mutant laminin is the N terminus, which is the region used to form a network. From this, one would predict that inclusion of any mutant laminin alpha2 would negatively affect the structural integrity of the basement membrane.
However, despite these concerns, the data show that the laminin α1 chain transgene ameliorated the dystrophic phenotype, restored muscle strength and reduced peripheral neuropathy showing that this technique might work for a wider cohort of patients.
You can read the older paper on lama1 overexpression to rescue the full lama2 knockout here and their follow-up with older animals here
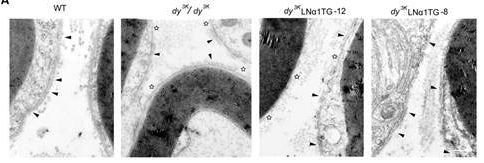
What’s next? It will certainly be interesting to see in these specific mice whether the effects are long lasting: are they truly cured or does the small amount of mutant laminin mean that the disease is just less severe? Delaying onset or milder phenotypes would still be incredibly useful!
These studies are proof-of-concept rather than a direct therapy at this point. Basically, they show that if we could induce laminin alpha1 expression in the muscles/nerves of human patients then their disease might be less severe. i.e. you don’t necessarily need to fix the mutation to get an effective therapy. An additional potential advantage is that you are inducing expression of a protein that already exists in patients therefore there should be less issues with potential immune responses to a new protein being made.
So the question now is, how can you increase laminin alpha1 expression and can you do it specifically? There are three or maybe four options that are being explored in a variety of contexts.
- Stop/slow its degradation at the mRNA level likely by blocking miRNA binding
- Turn on/up expression at the transcript level
- Introduce mRNA to encode for the protein eg via adenoviral delivery
- Ex vivo modification of cells to overexpress lama1, then transplant (though this might be really tough in these patients)
Each of these have challenges associated with them in terms of delivery, specificity and longevity of approach, so it will be interesting to see what comes next.
Linker Protein Therapy
Chimeric protein identification of dystrophic, Pierson and other laminin polymerization residues McKee et al, Matrix Biology
What’s the approach? Again this is a follow-up paper, building on previous work from Karen McKee published last year in Journal of Clinical Investigation. Here the principle is that by exploiting fundamental knowledge about laminin biology and laminin-binding proteins that a chimeric protein can be designed to bypass the problem that causes the disease phenotype.
Some of the Pierson syndrome and muscular dystrophy disorders occur because of a failure to form laminin networks rather than the mutated protein being not expressed at all? Well, in these cases the inherited missense mutations usually lie within the domain through which laminin polymer formation occurs, the LN domain. Here the authors have made a construct that contains a functional LN domain and fused it to part of the nidogen protein that binds to laminins. When this protein is expressed it localizes to laminins via the nidogen domain and stabilises laminin polymer formation via its LN domain. Super cool, super clever.
In the most recent article, mutations have been introduced into these chimeric proteins in order to determine which of those mutations affect laminin polymerisation. Knowing these details is important to fundamental biology but also will be information that can be used to determine which patients would benefit from this chimera protein therapy.
One of the reasons why I really like this work is that this sort of idea could not have come from a standard drug discovery pipeline. The concept of engineering a chimeric protein to have the features you need from two genetically distinct proteins comes from discovery science in the lab. There is a drive, especially in the UK, to focus only on research that has direct translational potential, at the expense of longer term, deeper mechanistic work. Using the current funding approach this sort of work would likely never have been supported.

What’s next? As with the other therapeutic approaches, the challenge is really how to get the protein in, and how to get it to the right place. The idea behind using a linker protein is that you can engineer this to be relatively small (1/10th the size of the whole protein therapy described above) and this should make things a lot easier in terms of recombinant production, purification in GMP facilities etc.
Conclusions
I think that these three papers, along with the work that led up to them, are really encouraging signs that things are moving in the right direction in terms of laminin genetic disease therapy. There is certainly hope that one or more of these approaches will continue to show promise and move toward the clinic.
Note: If you got this far and are surprised that I haven’t spoken about repairing the mutation or doing an ex vivo graft of repaired cells, then don’t be! That’s an option too! I wanted to focus on alternative approaches here. I did however, wrote a short piece on a ex vivo repair paper last year, you can read that here.
Comments/ideas for the next laminin research highlights post are most welcome (leave a reply below). Follow the blog or sign up for updates via email via the button.

1 Comment